1. Beneficial Effect of Inflammation in Obesity
Our research has changed view about the role of inflammation in obesity and type 2 diabetes. Chronic inflammation is associated with obesity and proposed as a cause of insulin resistance. However, it is not clear why the inflammation occurs and if it is a good therapeutic target in type 2 diabetes. Our study is designed to address these questions in animal and cellular models. Our result suggests that in obesity, the chronic inflammation is a feedback response to energy surplus with an important role to restore the energy homeostasis. This possibility is supported by our observations about initiation and impact of the inflammation in a series of studies. In 2007, we reported existence of adipose tissue hypoxia and its contribution to the obesity-associated chronic inflammation (1). In 2008, we published a report that the inflammation has a beneficial activity in promoting angiogenesis in adipose tissue (2). In 2010, we reported that adipose inflammation is able to stimulate energy expenditure in aP2-p65 mice to protect insulin sensitivity (3), the first report about disassociation of inflammation and insulin resistance in the field. Those findings about beneficial effects of inflammation lead us to propose that the chronic inflammation is protective feedback response in obesity (4). It is not a good target in the treatment of insulin resistance. This view provides a perfect answer to the disappointed results in most clinical trials of anti-inflammatory therapies for insulin resistance patients. In 2013, we published the first review article in the field to document evidence about the beneficial effects of obesity-associated inflammation (5).
2. New Approaches in Insulin Sensitization
We have identified a couple of new approaches in the treatment of type 2 diabetes. In addition to the inflammation study, we investigated approaches of improving insulin sensitivity. We reported that histone deacetylase inhibitor (sodium butyrate and TSA) stimulates energy expenditure in mice through an epigenetic mechanism (6), which provides a molecular mechanism for the beneficial effects of dietary fibers in the regulation of metabolism. We reported that herbal extract berberine regulates energy metabolism by transient inhibition of mitochondrial function (7), which leads us to propose that reversible mitochondrial inhibitors may be a new class of insulin sensitizers (8). We use the gastric bypass surgery model to study the mechanism of insulin resistance. We established the first mouse model of Roux-en Y gastric bypass (RYGB) that resembles human surgery in stomach size (9). We plan to study the mechanism of surgery using variety of transgenic mice. The works described here have been highly cited by colleagues. The total citation is above 7000 times according to data at “google scholar” on Jan. 20, 2014.
3. Adipose Tissue Hypoxia in Obesity
A. We demonstrated oxygen reduction in adipose tissue in obesity. Chronic inflammation occurs in adipose tissue and contributes to metabolic disorder in obesity. Several hypotheses have been proposed to explain the mechanisms, such FFA-TLR4, DAG-PKC, ER stress, ROS, and adipocyte death, et al. However, it is not clear what induces those risk factors? Our study provides an exciting mechanism for it. We demonstrate in fat tissue a reduction in the interstitial partial oxygen pressure (PO2), an increase in the hypoxia probe signal, and expression of the hypoxia response genes in ob/ob mice. The adipose hypoxia was confirmed in dietary obese mice as well. The hypoxia was associated with an increased expression of inflammatory genes and decreased expression of adiponectin. In dietary obese mice, reduction in body weight by calorie restriction was associated with an improvement in oxygenation and a reduction in inflammation. In cell culture, inflammatory cytokines were induced by hypoxia in primary adipocytes and macrophages of lean mice. The induction was associated with activation of transcription factors, NF-kB and HIF-1a. In addition, adiponectin expression was reduced by hypoxia through transcriptional inhibition in adipocytes. These data suggest a potential role of hypoxia in the induction of chronic inflammation and inhibition of adiponectin in the adipose tissue in obesity. These results were published in 2007 (1).
B. Our study suggests that an angiogenic failure may account for the hypoxia response in adipose tissue. In obesity, a reduction in blood supply in adipose tissue has been known for years. However, the biological significance was not clear. We believe that the hypoxia response is a result of the reduced blood supply. In search for the cause of blood supply reduction, we found that endothelial cell density reduced in the adipose tissue of ob/ob mice and macrophages were a signal amplifier of angiogenesis. The endothelial marker CD31 was reduced in protein and mRNA. Macrophage was found to be a major source of PDGF in adipose tissue. The macrophage PDGF was induced by hypoxia in cell culture. PDGF stimulated tube formation of endothelial cell. The PDGF activity was dependent on S6K. We conclude that in response to the reduced vascular density, macrophages express PDGF in adipose tissue to facilitate capillary formation in obesity. The capillary reduction is a result of lack of endothelial cells. The study suggests a new function of macrophages in the adipose tissue in obesity. This study was published in 2008 (2).
C. We reported that hypoxia inhibits adipocyte functions in several ways. We demonstrated that adipose tissue hypoxia is positively associated with body weight. The interstitial oxygen pressure (pO2) was reduced in the epididymal fat pads during weight gain. When body weight gained from 39.5 g to 55.5 g, pO2 declined from 34.8 mmHg to 20.1 mmHg, which are 40% - 60% lower than those in the lean mice. The hypoxia response impairs adipocyte function, and inhibits insulin signaling in the adipocytes. These effects are specific to adipocytes as they were only observed in adipocytes (3T3-L1), but not in myotubes (L6 myotubes). Hypoxia also reduces uptake of free fatty acid (FFA) by adipocytes through inhibition of fatty acid transporters (FATP1 and CD36), which inhibits adipocyte lipid accumulation. The mechanism is related to inhibition of transcription factor (PPAR and C/EBP) activities. The hypoxia also induces lipolysis, necrosis and apoptosis in adipocytes. These data suggest that adipose tissue hypoxia impairs adipocyte tissue function through direct suppression of adipocyte activities. This work was published in 2009 (10).
D. In an invited review article, we raised a new concept about adipose tissue hypoxia (ATH) in the adipose tissue dysfunction in obesity. ATH provides a cellular mechanism for adipose tissue dysfunction, such as chronic inflammation, macrophage infiltration, adiponectin reduction, leptin elevation, adipocyte death, ER stress and mitochondrial dysfunction in white adipose tissue in obesity (Fig. 1). The concept suggests that inhibition of adipogenesis and adipocyte function by hypoxia may be a new mechanism for elevated free fatty acids in the circulation in obesity. ATH may represent a unified cellular mechanism for variety of adipose tissue malfunctions, and insulin resistance in patients with metabolic syndrome. Additionally, it may help us to understand the beneficial effects of caloric restriction, physical exercise, and angiotensin II inhibitors in the improvement of insulin sensitivity. In this review, the evidence and possible cellular mechanisms of ATH are systemically analyzed, and the directions together with road blocks in the future studies are proposed. This review was published in 2009 (11).
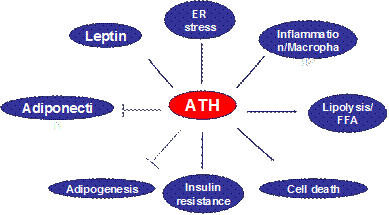
Fig 1. Impact of Adipose tissue hypoxia (ATH). ATH induces inflammatory response through activation of transcription factors such as NF-kB and HIF-1a. The activation may involve in ER stress or oxidative stress. ATH induces expression of leptin, cell apoptosis, lipolysis and insulin resistance. Persistent ATH also inhibits adiponectin expression and adipocyte differentiation.
E. We reported that inflammation serves a signal to stimulate energy expenditure in obesity. In addition to the study of inflammation origin, we also investigated biological significance of chronic inflammation in obesity. Using two different lines of transgenic mice, we demonstrated that inflammation serves as a signal to stimulate energy expenditure. The results were published in two JBC papers in 2009 – 2010 (3; 12).
4. Molecular Mechanism of Insulin resistance
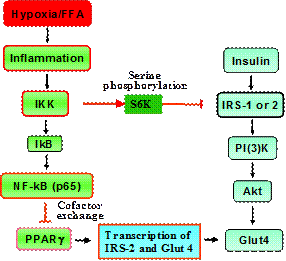
We explored molecular mechanism of insulin resistance in cellular models with a focus on two targets, IRS-1 and PPARg. Many studies including ours support that inflammation and free fatty acids (FFAs) are major pathogenic factors for insulin resistance in obese condition. We have been investigating the molecular mechanism by which inflammation and FFAs lead to insulin resistance. Our data suggests that inflammation, FFAs and hypoxia induce insulin resistance through activation of IKK/NF-kB signaling pathway as outlined in the following diagram.
Fig 2. Mechanisms of TNF/FFA-mediated Inhibition of Insulin Signaling Pathway in Adipocytes. TNF-a, FFAs and hypoxia lead to IKK activation through TNF-a receptor, TLR4 (Toll like receptor 4), and ROS, respectively. IKK induces insulin resistance by targeting IRS-1 and PPARg. IKK inhibits IRS-1 function through S6K-mediated serine phosphorylation at multiple sites such as S312/307 and S270/265. IKK inhibits PPARg function through activation of NF-kB. NF-kB is able to compete for the transcriptional coactivators or exchange corepressors with PPARg in the suppression of transcriptional activity of PPARg. This is responsible for inhibition of PPAR-target genes such as CAP and IRS-2. Our study shows that IKK promotes activity of nuclear corepressor of HDAC3 and SMRT. IKK induces nuclear translocation of HDAC3 from the cytoplasm. In the cytosol, HDAC3 associates with IkBa and degradation of IkBa promotes HDAC3 translocation into the nucleus. IKK acts by inducing IkBa degradation. Inhibition of PPARg activity leads to inhibition of insulin signaling by reducing IRS-2 expression, a signaling molecules in PI(3)k- dependent signaling pathways for Glut4 translocation.
A. FFA and IKK/NF-kB. Our study suggests that FFA may activates inflammation pathway through Toll-like receptor 4 (TLR4) (13). TLR4 is a receptor for bacteria endotoxin LPS and plays an important role in immune response to bacteria. TLR4 can be activated by non-bacterial agonists including saturated fatty acids. However, downstream signaling pathways for non-bacterial agonists are not known. Our study suggests that NF-kB was activated by saturated fatty acid (lauric acid) and the activation was inhibited by a dominant negative mutant of TLR4, MyD88, IRAK-1, TRAF6, or IkBa in macrophages (RAW264.7) and 293T cells. Lauric acid induced the transient phosphorylation of AKT. Inhibition of PI3K or AKT led to abolishment of NF-kB activation, p65 transactivation, and cyclooxygenase-2 (COX-2) expression in response to lauric acid. These results demonstrate that lauric acid is able to activate NF-kB and COX-2 expression partly through TLR4/PI3K/AKT/NF-kB signaling pathway. In contrast, docosahexaenoic acid (DHA) inhibited the phosphorylation of AKT induced by LPS or lauric acid. Together, these results suggest that saturated fatty acids use TLR4 receptor in the activation of NF-kB pathway. TLR4 may mediate FFA signals for inflammation response.
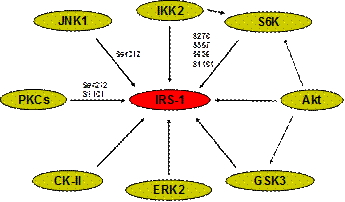
Fig. 3 Serine Kinases of IRS-1. Eight serine kinases have been identified for phosphorylation of IRS-1. IKK (Inhibitor kB Kinases) is a serine kinase in the control of transcription factor NF-kB activity. IKK activates NF-kB by releasing NF-kB from the inhibitor IkBa. This is finished by phosphorylation and degradation of IkBa. IKK catalyzes phosphorylation of IkBa on serine 32 and serine 36. JNK (JUN N-terminal Kinase) is a serine kinase in the MAPK family for activation of transcription factor c-JUN. c-JUN activation is marked by phosphorylation of serine 63 and serine 73 in c-JUN protein. Akt (PKB) is a serine kinase downstream of PI3K. Activation of Akt is marked by phosphorylation of Ser473 and Thr309 in Akt protein. GSK-3b (Glycogen Synthase Kinase 3 beta) is a kinase down stream of Akt that phosphorylates and inactivates GSK-3b. GSK-3b inactivates glycogen synthase through phosphorylation and thus promotes hydrolysis of glycogen. In response to insulin, GSK-3b is phosphorylated by Akt on serine 9 leading to glycogen biosynthesis. S6K (p70S6K) is a serine kinase downstream of mTOR and activated by Akt and other signals such as glucose and amino acids. PKCz (Protein Kinase C zeta) is a serine kinase parallel Akt in the PI3K signaling pathway. Activation of PKCz by insulin may contribute to GLUT4 translocation. ERK2 (extracellular signal-regulated kinase 2) is a member of the MAPK family. Activation of ERK is marked by rapid phosphorylation of threonine and tyrosine (Thr202/Tyr204) residues by its upstream kinase MEK1 (Map/Erk kinase-1). CK-II (Casein Kinase II) is a serine/threonine kinase that phosphorylates acidic protein such as casein. Though casein kinase 2 mainly phosphorylates rat IRS-1 on Thr502, and weakly on S99, the major site (Thr502) is not conserved between human and rodents.
B. IKK and IRS-1: We showed that the IKK complex phosphorylated IRS protein at serine residues including Ser312 in IRS-1 (Ser307 in rodents) (14). Recently, we reported that S6K1 (p70S6K) is involved in the IKK activity for IRS-1 serine phosphorylation (15). In the absence of S6K, inflammation was not able to induce insulin resistance. Insulin receptor substrate (IRS) is a protein that connects the insulin receptor to PI(3)K in the insulin signaling cascade. In the past few years, a consensus has emerged that serine phosphorylation of IRS-1 and IRS-2 contributes to the molecular mechanism of insulin resistance. Human IRS-1 contains more than 50 potential Ser/Thr phosphorylation sites. Phosphorylation at some of these Ser/Thr is inducible, and this inducible phosphorylation has been widely shown to lead to the functional inhibition of IRS-1. We observed that Ser312 phosphorylation led to the inhibition of IRS-1 association with the insulin receptor. The inducible Ser/Thr phosphorylation can be triggered by many risk factors of insulin resistance, including insulin, TNF-a, FFA, and inhibitors of serine phosphatase PP1 or PP2A (Calyculin A or Okadaic acid).
We observed that serine phosphorylation of IRS-1 has a biphasic effect on the function of IRS-1. In the early stage (with 3 hours), IRS-1 function is actually increased by the serine phosphorylation. An inhibition of IRS-1 association with PI(3)K was only observed after 3 hours. The molecular mechanism of this biphasic effect of serine phosphorylation is not clear. We believe that a sequential phosphorylation of IRS-1 by multiple serine kinases might be required for the inhibition. Cooperation among these kinases is likely to be critical.
Our data suggest that multiple serine kinases are activated by pathogenic factors of insulin resistance, such as TNF-a and FFAs (16; 17). The search for IRS-1 serine/threonine kinases has linked several serine kinases to IRS-1 (Fig. 2). In an effort to evaluate these serine kinases in TNF-induced insulin resistance, we examined six of these kinases (16). The results demonstrate that all of them (IKK, JNK, S6K, PKC, ERK and Akt) are activated by TNF-a, and activation of four kinases (IKK, JNK, mTOR and Akt) is blocked by aspirin. We observed that multiple kinases (such as IKK, JNK, PKC and mTOR) were also activated by FFAs in 3T3-L1 adipocytes, and activation of these kinases is associated with IRS-1 (Ser312/307) phosphorylation (17). Studies by our and other laboratories support that these FFA-responsive kinases contribute to Ser307/312 phosphorylation, however, a relationship of these serine kinases remains to be established.
C. IKK and PPARg: We and others have demonstrated that IKK inhibits the transcriptional activity of PPARg through activation of NF-kB. NF-kB is formed by two proteins of the Rel family, p65 and p50. The involvement of NF-kB in the transcriptional control of numerous cytokines and adhesion molecules has made NF-kB the most extensively studied transcription factor in the immune system. Using multiple systems, we demonstrated that activation of NF-kB led to inhibition of PPARg through several approaches (18). NF-kB inhibits mRNA expression of PPARg and this happens after overnight NF-kB activation. In acute manner, activation of NF-kB leads to inhibition of the transcriptional activity of PPARg through activation of HDAC3 (18). In this case, the DNA-binding activity of PPARg is not changed by NF-kB. Additionally, activation of IKK/NF-kB also promotes degradation of PPARg protein.
The transcriptional function of PPARg is required for the maintenance of insulin sensitivity. PPARg is a member of the peroxisome proliferator-activated receptors (PPARs) that include PPARa, PPARg, and PPARd (PPARb). PPARg function has been well established in the regulation of lipid and glucose metabolism. A dominant negative mutation of PPARg correlates to server insulin resistance. However, genetic mutations are not the major risk factors for PPARg dysfunction and PPARg function can be successfully restored by TZDs in large number of type 2 diabetes patients. This suggests that in most cases, the functional deficiency of PPARg is a result of regulatory failure. An understanding of this regulatory mechanism should lead to correction of the functional deficiency of PPARg. Our study has defined the possible mechanisms for the PPARg deficiency. Natural ligand of PPARg is believed to be the long chain fatty acids and their derivatives, such as 15dPGJ2 (15-deoxy 12-14- prostaglandin J2). This provides the physiological basis that FFAs have the potential to influence PPARa function. We believe that FFAs also can generate a negative feedback to inhibit PPARg function. Our study suggests that FFAs may use IKK/NF-kB pathway in the inhibition of PPARg function (13; 17).
Our study strongly support that the transcriptional coactivators and corepressors play a critical role in the inhibition of PPARg by NF-kB. In our study, function of PPARg is reduced by NF-kB or TNF-a (an activator of IKK/NF-kB) in the absence of loss of DNA-binding activity (18). The reduced PPARg activity is rescued by overexpression of any of the transcriptional coactivators including SRC-1, SRC-2 and p300. This suggests that the inhibition is a result of deprivation of the transcriptional coactivators from PPARg. The transcriptional activity of PPARg is dependent on recruitment of the transcriptional coactivators including SRC-1, SRC-2 and p300/CBP. SRC-1 (steroid receptor coactivator 1, NCoA-1) and SRC-2 (NCoA-2/TIF2/GRIP1) are two isoforms of the p160 protein. Interestingly, the same coactivators are also required by NF-kB. This is the molecular basis that NF-kB deprives PPARg of the transcriptional coactivators. Competition for a limited amount of the coactivators is a common mechanism of cross-inhibition between the two transcription factors. A good example is the inhibition of NF-kB transactivation by the glucocorticoid receptor. Although we have demonstrated this relationship between NF-kB and PPARg in a transcriptional model, it remains to be tested in the physiological condition.
5. Treatment of Insulin Resistance
In addition to the mechanistic study, we are also interested in treatment of insulin resistance with herbal medicines or dietary products. Prevalence of type 2 diabetes is around 7% in adult population in USA. The chemical drugs for type 2 diabetes treatment have variety of side effects (Table 1). Thiazolidinediones (TZDs) are the most effective anti-diabetes drug. Derivatives of TZD such as pioglitazone, rosiglitazone, englitazone, and ciglitazone are the most popular anti-diabetic drugs available to patients. TZDs enhance insulin sensitivity through activation of a nuclear receptor PPARg. However, application of TZDs is limited by the side effects (Table 1). Like TZDs, other synthetic anti-diabetic medicines also have severe side effects (Table 1). The side effects limit the application of chemical drugs, the demand for dietary supplement or alternative medicine has been increased dramatically in the care of insulin resistance and type 2 diabetes. Botanicals (herbal products) or functional food are the major ingredients in the hypoglycemic dietary supplement.
Table 1. Mechanisms and side effects of most common antidiabetic drugs |
||||
1 |
Name |
Examples |
Mechanism |
Side Effects |
2 |
Thiazolidinediones (TZDs) |
Pioglitazone Rosiglitazone |
Enhances insulin sensitivity |
Liver toxicity, weight gain, and CVD effects |
3 |
Biguanides |
Metformin |
Reduce hepatic gluconeogenesis |
Gastrointestinal disturbances |
4 |
Glucosidase inhibitor |
Acarbose |
Block glucose absorption |
Gastrointestinal disturbances |
5 |
Sulfonylureas |
Tolbutamide Glyburide |
Stimulate insulin secretion |
Hypoglycemia and weight gain |
6 |
Meglitinides |
Repadlinide |
Stimulate insulin secretion |
Hypoglycemia and weight gain |
7 |
Insulin |
|
Stimulate glucose deposition |
Hypoglycemia and weight gain |
Aspirin: It has been known for more than a century that salicylates (aspirin related chemicals) are able to reduce fasting blood glucose in diabetic patients. However, the cellular and molecular mechanism of the hypoglycemic activity of aspirin (salicylates) was not well elucidated. Several hypotheses were raised 20 years ago explaining the hypoglycemic action of aspirin. The extra-pancreatic hypothesis, which suggests that aspirin (salicylates) acts through a pancreatic-independent mechanism, seems to become dominant as it has gained substantial support from studies published recently.
We analyzed the action mechanism of aspirin in the regulation of insulin sensitivity in cellular models (16). Our study suggests that aspirin inhibits serine phosphorylation of IRS-1 induced by inflammatory cytokine TNF-a. In 3T3-L1 and Hep G2 cells, phosphorylation of IRS-1 at Ser307, Ser267, and Ser612 was induced by TNF-a. The phosphorylation was dependent on activation of IRS-1 serine kinases including IKK, JNK, Akt, ERK, mTOR, and PKCz. All of the six serine kinases were activated by TNF-a. Activation of four kinases like IKK, JNK, Akt and mTOR (but not ERK or PKCz) was inhibited by aspirin. This activity of aspirin led to prevention of serine phosphorylation of IRS-1 at Ser307, Ser267 and Ser612. Since these serine kinases (IKK, JNK, Akt and mTOR) are activated by many risk factors of insulin resistance, such as inflammatory cytokines, free fatty acids and free radicals (reactive oxygen species), this group of kinases may be involved in the pathogenesis of insulin resistance in obesity and aging. Inhibition of this group of seine kinases may contribute to the molecular mechanism by which aspirin enhances insulin sensitivity in the body.
Dietary products: Our study suggests that the short chain fatty acid may protect insulin sensitivity through induction of energy expenditure (6) and FGF21 is involved in the mechanism [Li, 2012 #8800]. Sodium butyrate, a salt of butyric acid, is a short chain fatty acid product that is found in cheese and diary products at 1-3%. It is also generated from dietary fiber after fermentation in the large intestine. We examined sodium butyrate in the regulation of insulin sensitivity in dietary obese mice. Dietary supplementation of butyrate prevented body weight gain without reducing food intake. It significantly increased energy expenditure in the mice. Adaptive thermogenesis and fatty acid oxidation were enhanced without an increase in spontaneous physical activity. Hepatic steatosis and adipose chronic inflammation were both reduced. Mitochondrial function was enhanced in brown adipose tissue and skeletal muscle with an elevation in mitochondrial biogenesis. PGC-1a expression was elevated at mRNA and protein levels together with mitochondrial genes (UCP-1, CPT-1b and COX-I). Most importantly, it attenuated insulin resistance in the mice. Taken collectively, the data suggest that butyrate improves insulin sensitivity through PGC1a-mediated energy expenditure and that sodium butyrate is a novel regulator of PGC-1α.
Botanicals (herbal medicine): We are interested in the metabolic activity of botanical products with special emphasis on the Chinese Herbal Medicine (19). We investigated the therapeutic effects of Berberine and its mechanism of action in the regulation of glucose metabolism (7; 20). Our study demonstrates that Berberine activates AMPK through inhibition of mitochondrial function (7). The inhibition leads to an increase in glucose utilization through glycolysis. Additionally, we investigated metabolic activity of Shilianhua, an herbal medicine. An extract of Shilianhua (F100) was able to reduced blood glucose in diabetic mice through improvement of insulin sensitivity (21).
| Table 2. Botanicals with hypoglycemic activities | ||||
| Plant Name | Form Tested | Effect | Side Effect | |
| 1 | Aloe vera | 80% juice | Decrease FBG | No effect on liver or kidney |
| 2 | Artocarpus | Boiled fresh leaves | Decrease PPG | Not reported |
| 3 | Coccinia indica | Powder of fresh leaves | Decrease FBG and PPG | No |
| 4 | Ginseng | Root powder | Decrease FBG and PPG | No effect on liver or kidney |
| 5 | Gymnema sylvestte | Extract | Decrease FBG | Not reported |
| 6 | Momordica charantia | Juice or protein extract | Decrease FBG and PPG | Hypersensitive reaction |
| 7 | Ocimum sanctum | Fresh leaf | Decrease FBG and PPG | No side effect |
| 8 | Opuntid streptacantha | Stem powder | Decrease FBG and insulin | Not reported |
| 9 | Silymarin (Milk Thistle) | Powder | Decrease FBG, insulin, urine glucose and C peptide | No side effect |
| 10 | Trigonclla foenum (Fenugreek) | Seed powder | Decrease FBG, PPG, insulin and urine glucose | No side effect |
Reference:
1. Ye J, Gao Z, Yin J, He H: Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of ob/ob and dietary obese mice. Am J Physiol Endocrinol Metab 2007;293:E1118-E1128
2. Pang C, Gao Z, Yin J, Zhang J, Jia W, Ye J: Macrophage Infiltration into Adipose Tissue May Promote Angiogenesis for Adipose Tissue Remodeling in Obesity. Am J Physiol Endocrinol Metab 2008;295:E313-E322
3. Tang T, Zhang J, Yin J, Staszkiewicz J, Gawronska-Kozak B, Mynatt R, Martin RJ, Keenan M, Gao Z, Ye J: Uncoupling of Inflammation and Insulin Resistance by NF-kB in Transgenic Mice through Induction of Energy Expenditure. J Biol Chem 2010;285:4637-4644
4. Ye J, Keller J: Regulation of energy metabolism by inflammation: A feedback response in obesity and calorie restriction. Aging 2010;2:361-368
5. Ye J, McGuinness OP: Inflammation during obesity is not all bad: Evidence from animal and human studies. Am J Physiol Endocrinol Metab 2013;304:E466-E477
6. Gao Z, Yin J, Zhang J, Pope CR, E WR, Martin RJ, Lefevre M, Cefalu WT, Ye J: Butyrate Improves Insulin Sensitivity and Increases Energy Expenditure in Mice. Diabetes 2009 58:1509-1517
7. Yin J, Gao Z, Liu D, Liu Z, Ye J: Berberine Improves Glucose Metabolism through Induction of Glycolysis. Am J Physiol Endocrinol Metab 2008;294:E148-E156
8. Zhang Y, Ye J: Mitochondrial inhibitor as a new class of insulin sensitizer. Acta Pharmaceutica Sinica B 2012;4:341-349
9. Hao Z, Zhao Z, Berthoud H-R, Ye J: Development and Verification of a Mouse Model for Roux-en-Y Gastric Bypass Surgery with a Small Gastric Pouch. PLoS ONE 2013;8:e52922
10. Yin J, Gao Z, He Q, Ye J: Role of hypoxia in obesity-induced disorders of glucose and lipid metabolism in adipose tissue. Am J Physiol Endocrinol Metab 2009;296:E333-E342
11. Ye J: Emerging Role of Adipose Tissue Hypoxia in Obesity and Insulin Resistance. Int J Obes 2009;33:54-66
12. Gao Z, Yin J, J. Z, He Q, McGuinness OP, Ye J: Inactivation of NF-κB p50 Leads to Insulin Sensitization in Liver through Post-translational Inhibition of p70S6K. J Biol Chem 2009;284:18368-18376
13. Lee JY, Ye J, Gao Z, Youn HS, Lee WH, Zhao L, Sizemore N, Hwang DH: Reciprocal modulation of Toll-like receptor-4 signaling pathways involving MyD88 and phosphatidylinositol 3-kinases/AKT by saturated and polyunsaturated fatty acids. J Biol Chem 2003;278:37041-37051
14. Gao Z, Hwang D, Bataille F, Lefevre M, York D, Quon MJ, Ye J: Serine phosphorylation of insulin receptor substrate 1 by inhibitor KappaB kinase complex. J Biol Chem 2002;277:48115-48121
15. Zhang J, Gao Z, Yin J, Quon MJ, Ye J: S6K Directly Phosphorylates IRS-1 on Ser-270 to Promote Insulin Resistance in Response to TNF-α Signaling Through IKK2. J Biol Chem 2008;283:35375-35382
16. Gao Z, Zuberi A, Quon M, Dong Z, Ye J: Aspirin Inhibits Serine Phosphorylation of Insulin Receptor Substrate 1 in Tumor Necrosis Factor-treated Cells through Targeting Multiple Serine Kinases. J Biol Chem 2003;278:24944-24950
17. Gao Z, Zhang X, Zuberi A, Hwang D, Quon MJ, Lefevre M, Ye J: Inhibition of Insulin Sensitivity by Free Fatty Acids Requires Activation of Multiple Serine Kinases in 3T3-L1 Adipocytes. Mol Endocrinol 2004;18:2024-2034
18. Gao Z, He Q, Peng B, Chiao PJ, Ye J: Regulation of Nuclear Translocation of HDAC3 by IkBa Is Required for Tumor Necrosis Factor Inhibition of Peroxisome Proliferator-activated Receptor {gamma} Function. J Biol Chem 2006;281:4540-4547
19. Yin J, Zhang H, Ye J: Traditional chinese medicine in treatment of metabolic syndrome. Endocr Metab Immune Disord Drug Targets 2008;8:99-111
20. Yin J, Xing H, Ye J: Efficacy of berberine in patients with type 2 diabetes mellitus. Metabolism 2008;57:712-717
21. Yin J, Zuberi A, Gao Z, Liu D, Liu Z, Ye J: Shilianhua extract inhibits GSK-3{beta} and promotes glucose metabolism. Am J Physiol Endocrinol Metab 2009;296:E1275-E1280